Possible Causes and Solutions to Common Sequencing Issues
The most common factors limiting sequence quality:
-
Impure template DNA
- Contaminants: salts (EDTA, NaCl, NaAc, Kac, KCl), proteins, detergents (SDS, Triton X-100), RNA, chromosomal DNA, organic chemicals (ethanol, chloroform, phenol), divalent cations (Mg, Ca, Mn), and excess PCR primers, dNTPs, enzyme, and buffer components from PCR. Be sure to check the quality of the DNA before submitting for sequencing.
- Incorrect template or primer concentration
- Sub-optimal primer selection or annealing
- Secondary structure of template
Some solutions from the Troubleshooting Guide to Sanger Sequencing are listed below. Contact Core staff for further help and information.
Good Sequencing
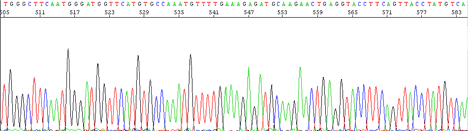
The sequencing electropherogram will show machine model and analysis software version in the left hand corner. The sample name and investigator will appear to the middle left. On the middle right you will see notation showing signal strength. Typical good signal will show GATC values 100-600.
Peaks are sharper for the first 500 bases, resolution of peaks drops off from that point, but are still readable out to 750 bases.
Bad and Not-So-Good Sequencing
The following shows some examples of problems in sequencing and what the possible reason for failure may be. This list is by no means complete or all-inclusive of reasons for failures. If you notice any obvious omissions or alternate solutions, please let us know.
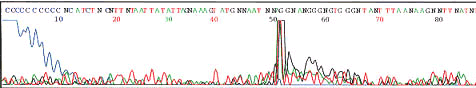
Complete failure - No signal present at all
| Possible Cause | Solution |
|---|---|
|
Notice poly C from dye front and one to two spikes
|
See section below on low signal strength for suggested solutions
|
|
Insufficient template
|
Quantitate the double stranded DNA by fluorimeter, Double check gel quantitation. Increase the amount of DNA
|
|
Inhibitor contaminating template such as salt or ethanol
|
Clean up template. Spin columns
|
|
Insufficient primer
|
Make sure you diluted primer to correct concentration. 12 pmol primer per reaction (NOT 12 picomolar)
|
|
Primer has no annealing site, poor primer design (low melting temp) or incorrect primer sequence
|
Pick good sequencing area. If there are any mismatches between the primer and template the primer will not anneal correctly. Do not make primer in areas of ambiguity.
|
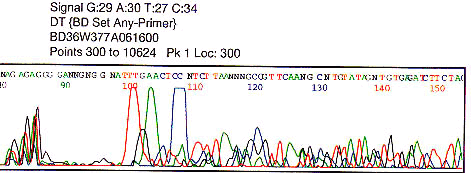
Low signal strength of G A T C values less than 50
| Possible Cause | Solution |
|---|---|
|
Insufficient template
|
Quantitate the double stranded DNA by fluorimeter, Double check gel quantitation. Increase the amount of DNA
|
|
Inhibitor contaminating template such as salt or ethanol
|
Clean up template. Spin columns
|
|
Insufficient primer
|
Make sure you diluted primer to correct concentration. 12 pmol primer per reaction (NOT 12 picomolar)
|
|
Primer has no annealing site, poor primer design (low melting temp) or incorrect primer sequence
|
Pick good sequencing area. If there are any mismatches between the primer and template the primer will not anneal correctly. Do not make primer in areas of ambiguity.
|
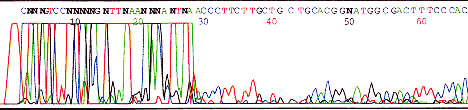
Primer-dimer
| Possible Cause | Solution |
|---|---|
|
Primer dimer caused by self binding primer or primer binding to other primers present in template mixture (PCR). Short fragment is predominately amplified. There may or may not be any sequencing following such an artifact.
|
Analyze primer used for sequencing.
Could template be contaminated with other primers to cause primer dimers? Is point of drop-off at the cloning site? |
Abrupt signal loss

Possible Cause Solution
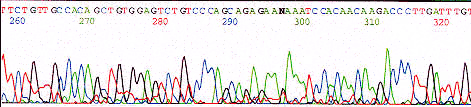
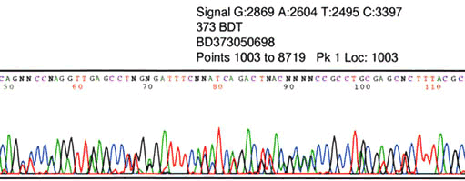
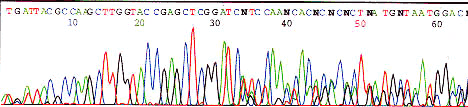


PCR product
This is normal
Plasmid insert with secondary structure
Sequence opposite strand, try a primer that anneals in a different position. Ask Core Labs staff to add DMSO or try other techniques
Noisy Sequencing
Probable cause: Low signal strength due to low template/primer or impurities such as salt or ethanol
Probable cause: Multiple templates/primers/priming sites or high signal strength
Probable cause: Mixed plasmid prep, two inserts
| Possible Cause | Solution |
|---|---|
|
low signal strength
|
Low signal strength may cause background to appear even higher. Check for impurities. Increase primer and/or template concentration
|
|
high signal strength (saturating detector)
|
Decrease template concentration
|
|
Contaminated template (impurities)
|
Purify template with spin columns
|
|
Mixed plasmid prep, vector portion is clean 2 inserts seen
|
streak for isolation 2X
|
|
Multiple templates/primers/priming sites
|
Streak plasmids for isolation 2X,
Are there possible multiple priming sites? N-1 primer? If a PCR product make sure you removed original primers, also one primer may have generated both ends, or primers may amplify more than one product of DNA. |
Slow drop-off read / Early signal loss
| Possible Cause | Solution |
|---|---|
|
Low template or primer concentration
|
Increase either primer or template concentration
|
|
Contamination with salt, ethanol, or other impurity
|
Purify template with spin columns
|
|
High GC content
|
See next section
|
|
High GT content
|
Talk to Core Facilities staff about what we can do
|
|
Secondary structure
|
Sequence opposite strand, try a primer that anneals in a different position. Ask Core Labs staff to add DMSO or try other techniques
|
High GC content with short read
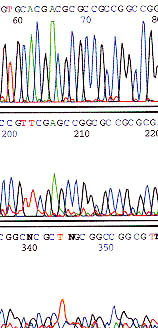
Talk to Core Facilities staff to determine alternate sequencing strategies such as adding DMSO, increasing denaturation temperatures, alternate sequencing chemistries
Waves due to enzyme slippage at poly T region
| Possible Cause | Solution |
|---|---|
|
The DNA polymerase may slip during sequencing reaction when it reaches a poly base region. A partially synthesized strand will rebind a base ahead or behind where it should and a series of products are generated with slightly different lengths of poly base regions. This generates a wave pattern as seen above.
|
Use an anchored primer or alternate sequencing chemistry such as dRhodamine.
|